Catalysis · DFT · Machine Learning
2026
Catalyst Activity Predictor
Predicting which catalyst works before running the reaction.
The simulation engine I built for Reformix could tell me whether a given vitrimer formulation had the right glass transition temperature and the right mechanical properties. What it couldn't tell me, not directly, was how fast the bond exchange reaction would happen. That's a separate question, and it turned out to be just as important.
The speed of bond exchange in a vitrimer determines everything about its practical behavior. Too slow, and the material takes hours to remold at elevated temperature, which makes it useless for manufacturing. Too fast, and stress relaxation happens at room temperature, and the material creeps under load and can't hold its shape. The target is a specific window: exchange timescales of seconds to minutes at 120 to 180°C, and effectively zero exchange at room temperature. The catalyst is the primary lever for tuning where that window sits.
Choosing the right catalyst by trial and error means months of synthesis and testing. I wanted to compute it instead.
What the catalyst actually does
In ester-based vitrimers, the bond exchange mechanism is transesterification: an alcohol attacks an ester carbonyl, forms a tetrahedral intermediate, and one of the original ester's oxygen-carbon bonds breaks while a new one forms. The net result is that the ester group has migrated from one alcohol partner to another. The network topology changes without any net change in the number of bonds, which is why vitrimers are insoluble at all temperatures: the network never opens up completely, it just reshuffles.
Without a catalyst, this reaction has an activation barrier high enough that it's negligibly slow below about 200°C. The catalyst lowers that barrier. The question is by how much, and different catalyst chemistries lower it in different ways and by different amounts.
Zinc acetate, the most common catalyst in the vitrimer literature, operates by coordinating to the ester carbonyl oxygen and activating it toward nucleophilic attack. Tertiary amine bases like DMAP and TBD operate differently: they activate the attacking alcohol by deprotonating it or by acting as nucleophilic co-catalysts that form acyl-imidazolium intermediates. Brønsted acid catalysts like p-toluenesulfonic acid protonate the carbonyl oxygen directly. Each of these mechanisms implies a different transition state geometry and a different set of electronic properties that should correlate with catalytic activity.
The question I was asking is: given a molecular structure for a candidate catalyst, can you predict the activation barrier for the transesterification reaction it catalyzes, without running a full transition state search in DFT?
Building the dataset
This project started with a literature mining effort. I needed activation barriers for transesterification reactions catalyzed by a range of chemically diverse catalysts, where both the catalyst structure and the activation barrier had been reported in the same study or could be cross-referenced.
The literature on organocatalytic transesterification is large but scattered. Most papers report reaction rates or yields, not activation barriers directly. Converting between rate constants and activation barriers via the Eyring equation requires also knowing whether the rate is the intrinsic chemical step or a diffusion-limited or mass-transfer-limited observable. I restricted the dataset to studies that either reported free energies of activation from Eyring analysis of temperature-dependent rate data, or that reported DFT-computed barriers for the specific mechanism that was experimentally verified.
This filtering left me with 94 catalyst-substrate-barrier data points across 31 distinct catalyst structures. That's not large by machine learning standards. But the features I planned to use were not raw molecular graphs: they were DFT-derived electronic descriptors that compress a lot of information into a small number of meaningful numbers, which means the effective dimensionality of the feature space is manageable even with limited data.
Computing the descriptors
For each of the 31 catalyst structures, I ran DFT calculations in ORCA using the same protocol I had established for the NNP training project: geometry optimization at B3LYP/def2-SVP, followed by single-point energy at B3LYP/def2-TZVP with the D3BJ dispersion correction, followed by natural bond orbital (NBO) analysis.
The descriptors I extracted fell into three categories.
Electronic descriptors: HOMO and LUMO energies, chemical hardness (half the HOMO-LUMO gap), the Fukui function evaluated at the catalytically active atom (a measure of local electrophilicity or nucleophilicity depending on direction), and the natural charge on the key donor or acceptor atom. For Lewis acid catalysts like zinc acetate, the relevant atom is the metal center. For nucleophilic catalysts like DMAP, it's the nitrogen. For Brønsted acids, it's the proton-donating oxygen.
Steric descriptors: the buried volume around the active site, computed using the Sterimol parameters of the substituents, and the solvent-accessible surface area of the active pocket. Steric effects on transesterification are real: a very bulky catalyst that coordinates to the carbonyl might lower the intrinsic barrier but also block the incoming nucleophile.
Thermodynamic descriptors: the binding energy of the catalyst to a model ester substrate in its ground state configuration, and the proton affinity of the catalyst's basic site where applicable. These capture something about how strongly the catalyst interacts with the substrate before the transition state, which through the Bell-Evans-Polanyi principle correlates with the transition state energy.
Computing all of these for 31 catalyst structures took about two weeks of intermittent GPU time. Most of the calculations ran cleanly. Four required manual intervention because the initial geometry guesses produced structures that optimized to incorrect conformations, which showed up as imaginary frequencies in the vibrational analysis that didn't correspond to the reaction coordinate.
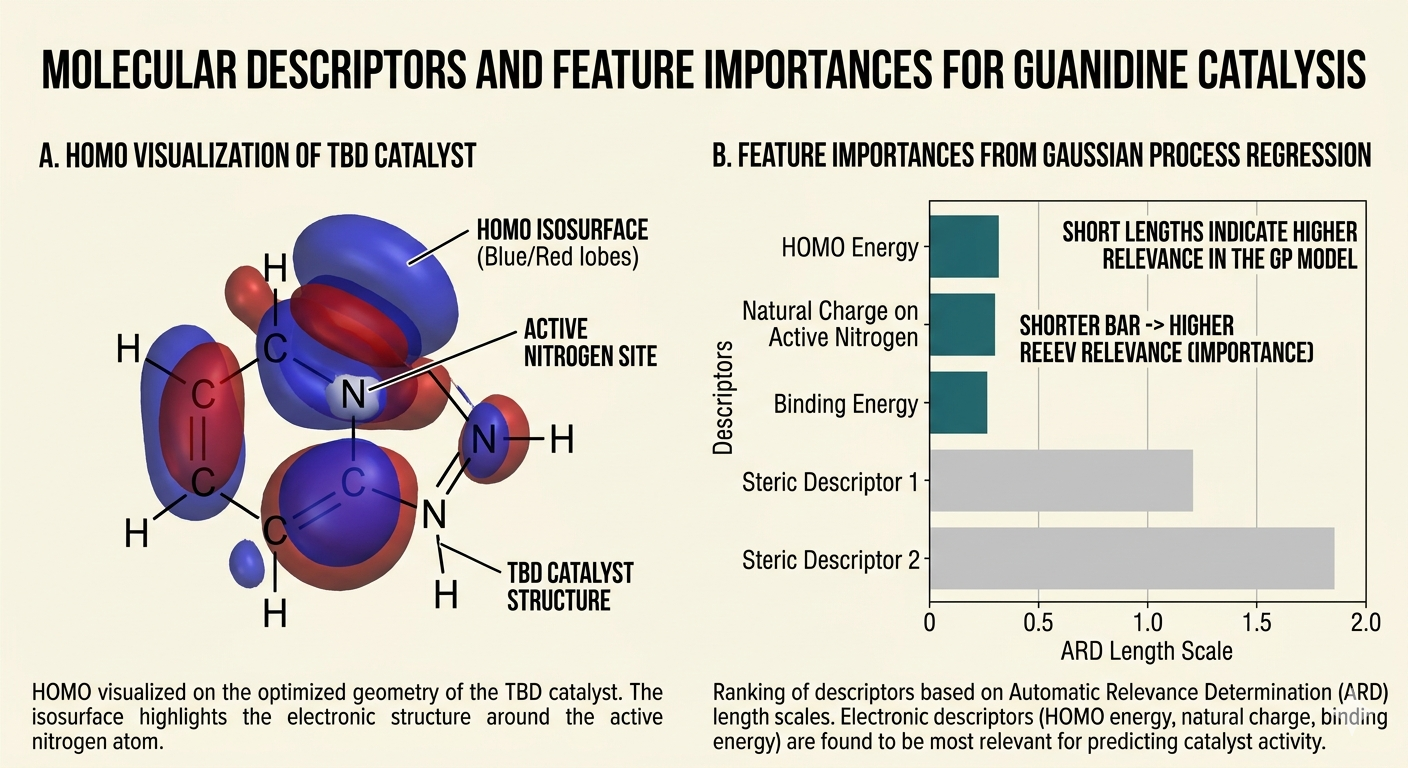
The model
With 94 data points and around 15 descriptors per catalyst, I had a choice between simple regression models and more expressive ones. With this data size, complex models are likely to overfit unless carefully regularized.
I tried three approaches in parallel. The first was a Gaussian Process regression directly on the descriptor vector, using an ARD (automatic relevance determination) kernel that learns separate length scales for each descriptor, effectively performing implicit feature selection. The second was a gradient-boosted tree ensemble (XGBoost), which handles nonlinear interactions between descriptors naturally and has built-in regularization. The third was a graph neural network operating directly on the molecular graph of the catalyst, using message passing to learn a molecular embedding and then predicting the activation barrier from the embedding.
The GNN approach was the one I was most conceptually interested in because it doesn't require manual descriptor design. You give it the molecular graph and it learns what matters. In practice, with 94 data points, it was also the approach most prone to overfitting. I ran it with heavy dropout and early stopping based on a held-out validation set, which helped, but the performance was not better than the GP with hand-crafted descriptors despite using orders of magnitude more parameters.
The GP with ARD kernel performed best in cross-validation, with a mean absolute error of about 4.2 kJ/mol on held-out data. For context, the total range of activation barriers in the dataset was around 60 kJ/mol, so 4.2 kJ/mol represents reasonable but not extraordinary accuracy. Enough to rank catalyst families reliably. Not accurate enough to make confident quantitative predictions for any individual catalyst without experimental validation.
The ARD length scales revealed which descriptors mattered most. The HOMO energy of the catalyst, the natural charge on the active site atom, and the binding energy to the model substrate were the three features with the smallest length scales, meaning the model was most sensitive to variation in these quantities. Steric descriptors mattered much less for the reaction systems in my dataset, most of which used small model substrates rather than bulky polymer chain fragments. That's a limitation: steric effects might become more important when the substrate is a polymer chain with restricted mobility, and the model might not transfer accurately to that regime.
Predicting new catalysts
With a trained model and calibrated uncertainty estimates from the GP, I generated predictions for 200 catalyst candidates. These came from two sources: computationally enumerated analogs of the best-performing catalysts in the training set (varying substituents systematically), and a subset of the molecules generated by the VAE from the Generative Molecule Designer that had appropriate functional groups to serve as organocatalysts.
The predictions flagged 18 candidates with predicted barriers more than 8 kJ/mol below the best catalyst in the training set, which is a meaningful reduction. Eight of these were analogs of TBD with electron-donating substituents on the guanidine core. The rationale is clear from the descriptor analysis: TBD works by activating the attacking nucleophile through hydrogen bond donation, and electron-donating groups increase the nitrogen nucleophilicity and the N-H acidity simultaneously, both of which should lower the barrier. The model is essentially learning and extrapolating this chemical intuition, which is reassuring: it's not finding spurious correlations, it's recovering the mechanism.
Four of the 18 flagged candidates were not analogs of known catalysts but novel structures from the generative model. These I treated with more skepticism, because the descriptor extrapolation from the training distribution to these structures is larger and the uncertainty estimates correspondingly wider. I computed full DFT transition state searches for these four using a simplified model substrate (ethyl acetate plus methanol) to get a ground-truth comparison. Two of the four showed barriers within 5 kJ/mol of the model prediction. One showed a barrier 12 kJ/mol higher than predicted, which is a significant miss and corresponds to a steric effect the descriptor set didn't capture adequately. One failed to converge to a transition state using standard methods, suggesting the reaction mechanism for that catalyst is more complex than assumed.
That 50% quantitative accuracy on novel scaffolds is not impressive as a standalone number. What it means in practice is that the model is a useful pre-filter, not a replacement for DFT. It can rank 200 candidates and identify a shortlist of 18 worth running DFT on, rather than running DFT on all 200. That's still a significant acceleration of the search process.
The thread through all of it
This project sits at the end of a chain. The Reformix simulation engine established that you can compute vitrimer properties from first principles. The NNP trainer made that computation faster. The active learning loop made the search smarter. The generative model produced novel molecular candidates. This project closes the loop by predicting whether those candidates can be catalyzed efficiently, because a vitrimer formulation that can't be processed at practical temperatures is useless regardless of how good its mechanical properties look in simulation.
The full pipeline, all nine projects together, is a computational infrastructure for designing recyclable materials from scratch. Each piece is independently useful. Together they describe a research methodology: generate candidates, screen by properties, validate the chemistry, optimize the processing. The common thread is that every step is guided by computation before it's validated by experiment, which inverts the traditional order of trial-and-error synthesis followed by retrospective understanding.
Whether that inversion is faster in practice depends on how accurate the computations are, and that depends on continued validation against experiment. Every prediction in these projects is a hypothesis. The experiments that will test them are the next chapter.