Climate · Porous Materials · Simulation
2025
CO2 Capture MOF Screener
Searching 10,000 crystal structures for the right CO2 trap.
There are currently around 422 parts per million of CO2 in the atmosphere. That number is going up by about 2.5 ppm every year. Every credible scenario for staying below 2°C of warming includes not just cutting emissions but actively removing CO2 that's already up there. The problem is that air is enormous and CO2 in it is thin. Pulling a specific molecule out of a vast, well-mixed gas mixture at 0.04% concentration requires a material that is exceptionally good at discriminating between CO2 and everything else, binding it selectively and reversibly, and doing it fast enough and cheaply enough to matter at the scale of gigatons per year.
The class of materials best positioned to do this is metal-organic frameworks. I built a computational pipeline to search through them.
What a MOF is and why it matters here
A metal-organic framework is a crystalline solid made by coordinating metal ions or clusters with organic linker molecules into a three-dimensional lattice. The result is a structure that is almost entirely empty space, which is the point. MOFs have internal surface areas that sound made-up until you've thought about them carefully: some reach 7,000 square meters per gram. A single gram of the right MOF has more internal surface area than a soccer field. All that surface is available for gas molecules to adsorb onto, and the geometry and chemistry of the pores determines which gas molecules adsorb preferentially.
That selectivity is what makes MOFs interesting for CO2 capture specifically. CO2 has a large quadrupole moment, which means it interacts favorably with polar pore environments. N2 and O2, which make up most of the air you're trying to separate it from, have much weaker interactions with most surfaces. A MOF with the right combination of pore size, shape, and surface chemistry can bind CO2 selectively while leaving the rest of the air largely indifferent.
The catch: there are over 100,000 MOFs that have been synthesized and characterized, and the CoRE MOF database, which is the curated subset with high-quality crystal structures suitable for computation, still contains around 14,000 entries. No experimental group is going to test 14,000 materials. You need computation to pre-screen.
The simulation approach
Adsorption in porous materials is governed by statistical mechanics. At equilibrium, the amount of gas adsorbed at a given temperature and pressure is determined by the balance between the energetic favorability of adsorption and the entropic cost of confining gas molecules to specific sites. The simulation technique that captures this directly is Grand Canonical Monte Carlo (GCMC).
In GCMC, you hold temperature and chemical potential fixed (which fixes pressure) and allow the number of gas molecules in the simulation box to fluctuate. Molecules are randomly inserted, deleted, and moved according to Metropolis acceptance criteria. After enough steps, the system equilibrates and the average number of adsorbed molecules gives you the adsorption isotherm. Do this across a range of pressures and you have the full adsorption curve.
I implemented this in RASPA2, which is the standard open-source code for this class of simulation. The force field for CO2 is TraPPE (Transferable Potentials for Phase Equilibria), which describes CO2 as a rigid three-site linear molecule with partial charges and Lennard-Jones parameters that reproduce the experimental equation of state accurately. Framework atoms use the UFF force field with partial charges derived from the DDEC charge equilibration method applied to the crystal structure.
The key outputs I extract from each simulation: CO2 uptake at working conditions: specifically the uptake at 0.15 bar CO2 partial pressure (post-combustion flue gas conditions) and at 0.4 mbar (direct air capture conditions). Selectivity over N2: I run GCMC for pure CO2 and pure N2 at their respective partial pressures and use Ideal Adsorbed Solution Theory (IAST) to estimate the mixture selectivity. Working capacity: the difference in CO2 uptake between adsorption pressure and desorption pressure. I flag materials where the isosteric heat of adsorption is above 60 kJ/mol. Henry's law constant at low pressure: the initial slope of the adsorption isotherm — the most relevant metric for DAC, where you're working in the linear regime.
What I found screening the CoRE MOF database
The screening took several weeks of compute time spread across batches. The distribution of performance is heavily skewed. Most MOFs are mediocre for CO2 capture. A significant fraction are actively bad, either because their pores are too small for CO2 to enter, or because they adsorb everything indiscriminately with no selectivity, or because they have dangling bonds that collapse the structure when you apply charge equilibration.
The pre-filter I developed checks four things: minimum pore diameter above the kinetic diameter of CO2 (3.3 Å), accessible surface area above zero, no partial charges that violate electroneutrality after charge equilibration, and structural stability under the force field. This filter removed about 30% of the database before GCMC even started.
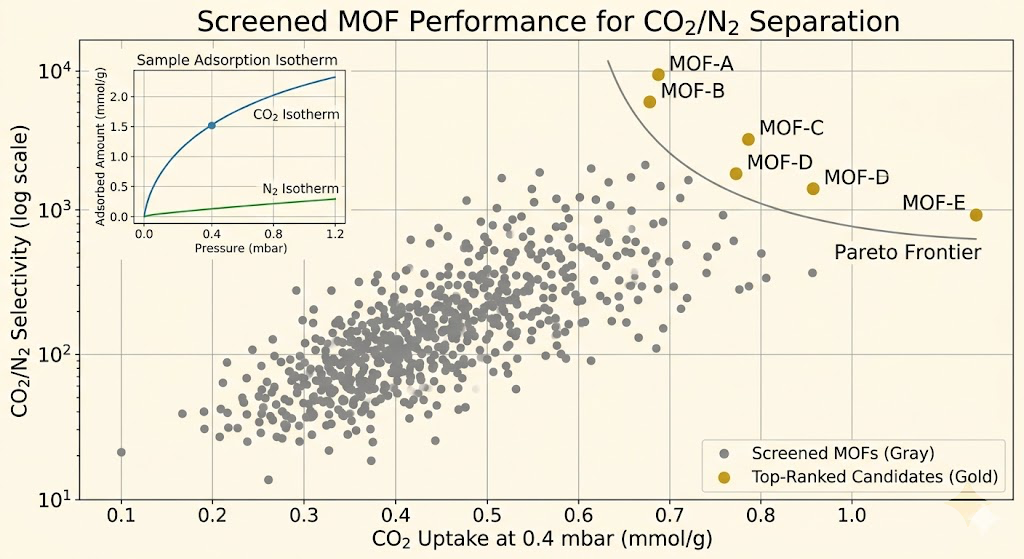
Of the remaining structures, roughly 200 showed selectivity above 100 for CO2/N2 under post-combustion conditions. That's 1.4% of the database. Of those 200, about 40 also showed working capacities above 1 mmol/g across a reasonable pressure swing. Of those 40, about a third had isosteric heats of adsorption below 60 kJ/mol, meaning thermal regeneration should be energetically reasonable.
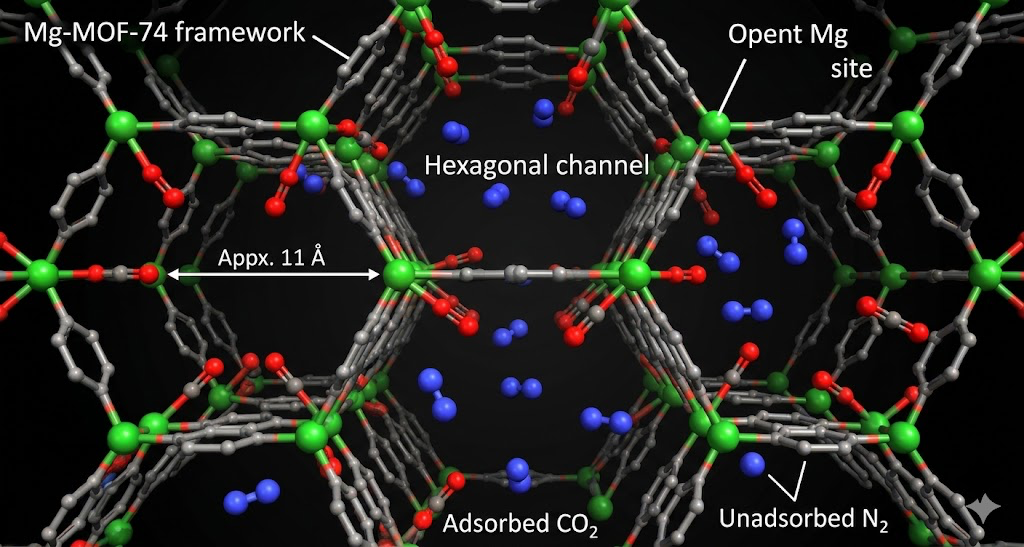
The structures that scored best clustered around specific metal nodes and linker chemistries. Frameworks with open metal sites, coordinatively unsaturated metal centers that can directly coordinate the oxygen of CO2, consistently showed the highest Henry's law constants at low pressure. Mg-MOF-74 kept showing up near the top for DAC conditions specifically because of how strongly the Mg²⁺ open metal site interacts with CO2's oxygen.
The DAC challenge specifically
Direct air capture is a fundamentally harder problem than post-combustion capture, and the screening results make this concrete. At 0.4 mbar CO2 partial pressure, the overwhelming majority of MOFs show negligible uptake. You need materials with Henry's law constants above roughly 100 mmol/g/bar to get meaningful working capacity at air concentration.
Only a handful of structures in the CoRE MOF database hit that threshold without also having regeneration energetics that make them impractical. The open metal site frameworks are the main candidates, and even they have issues: Mg-MOF-74 is sensitive to water, which is present in ambient air and competes with CO2 for the same sites. The computational screening doesn't directly model water competitive adsorption, so I added a literature flag for water stability for every top-ranked structure.
This is one of the places where the computational pipeline runs into the limits of what simulation can tell you without experimental validation. The force fields for water in MOFs are worse than for CO2. The modeling of competitive adsorption from humid air is a research problem in itself. The screening identifies promising candidates, but the water stability question requires experimental measurement or much more expensive AIMD simulations of the specific adsorption event.
What the pipeline produces and what it doesn't
The pipeline produces a ranked list of candidates with quantitative predictions for CO2 uptake, CO2/N2 selectivity, working capacity, and isosteric heat of adsorption across the full CoRE MOF database. It produces uncertainty estimates based on sensitivity analysis of the force field parameters. It flags structures where the geometry optimization produced unusual configurations, where the charge equilibration converged slowly, or where the GCMC uptake at adjacent pressure points is non-monotonic.
What it doesn't produce is a synthesis route. The database contains experimentally synthesized structures, but synthesizing a specific MOF reliably, at scale, with consistent pore geometry and no defects, is a separate craft. Some of the top-ranked structures in the database are known to be difficult to reproduce reliably. I flagged those based on literature reports.
The project produced a clear shortlist, a set of structural features associated with good DAC performance, and a reproducible pipeline that can be re-run as new crystal structures are added to the database. The CoRE MOF database is updated roughly annually. Running the pipeline on each new version is straightforward, which means the shortlist stays current.
The broader point is what this kind of computational pre-screening does to the research process. Experimental groups working on CO2 capture materials are not short on creativity. What they're short on is time and synthesis resources. A list of 40 validated computational candidates with explicit predictions for each one changes what questions they spend their time on. The synthesis question becomes: of these 40, which are already known to be synthesizable reliably, which are close analogs of known structures, and which are genuinely novel? That triage is faster and more rigorous than intuition alone.