Generative AI · Molecular Design · Computational Chemistry
2026 — ongoing
Inverse Molecular Design Engine
Start from the property. Work backward to the molecule.
Most computational tools in materials science work in one direction. You have a molecule, you simulate it, you get its properties. Density, glass transition temperature, modulus, conductivity, whatever you care about. This is useful because it is faster than synthesizing the molecule and measuring it in a lab. But it still assumes you already know what molecules to look at. You still have to propose candidates manually, or search through formulation space by hand, and then run simulations to evaluate each one.
I wanted to flip the direction entirely. Tell the system what properties you want. Get back the molecular structure that achieves them.
This is called inverse design, and it is genuinely hard in a way that forward simulation is not. Forward simulation is a function. One input, one output, and the function is smooth and well-behaved enough that classical mechanics handles it well. Inverse design is not a function. Many different molecular structures can have similar glass transition temperatures. The mapping from properties back to structures is one-to-many, and many-to-many, and the landscape is full of discontinuities because small changes in molecular structure can produce large changes in macroscopic behavior. You cannot just invert the forward model.
The approach that works is generative. You train a model that learns the joint distribution over molecular structures and their properties, and then you condition the generation process on your target properties. Sample from that conditional distribution and you get candidate molecules that the model believes will meet your specifications.
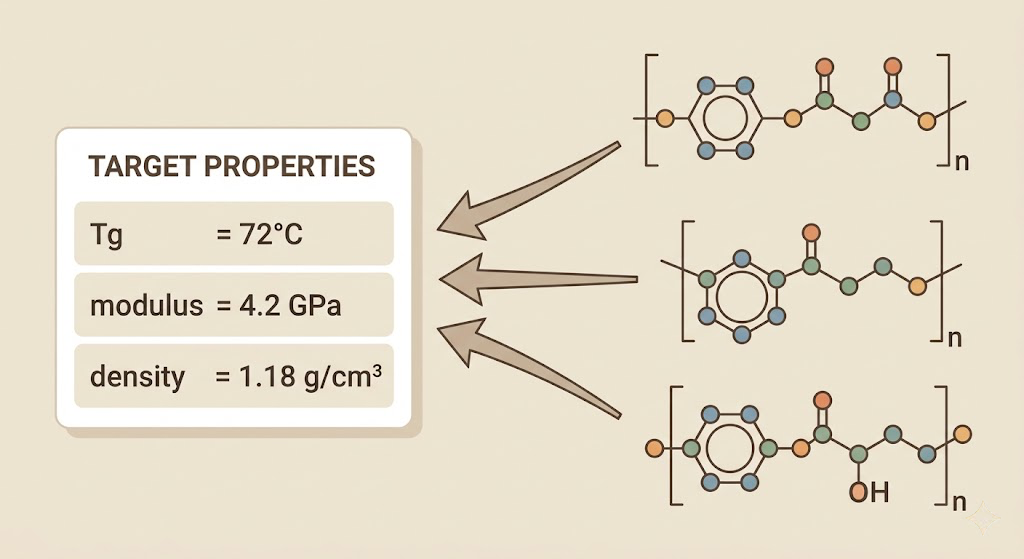
The starting point for this project was the dataset I had already built for the NNP trainer: thousands of molecular configurations with DFT-computed energies and forces, and thermomechanical properties computed via MD for the validated systems. I extended this significantly by pulling from three public databases. The PolyInfo database contains experimentally measured properties for several thousand polymers. The Harvard Organic Photovoltaic Dataset has 26,000 molecules with computed properties. The QM9 dataset has 134,000 small organic molecules with DFT properties. None of these are vitrimer systems specifically, but the model does not need to know about vitrimers. It needs to learn the general relationship between molecular structure and macroscopic behavior, and the vitrimer-specific data from Reformix gives it supervised signal on the properties I actually care about.
Representing molecules in a way a neural network can work with is its own problem. SMILES strings are the standard text representation, one character per atom or bond, and they have the advantage of being compact and human-readable. The disadvantage is that small edits to a SMILES string often produce invalid molecules or completely different chemistry, which makes them hard to use as a latent space for generation. Molecular graphs are better for learning because they encode the actual connectivity structure, with atoms as nodes and bonds as edges, and graph neural networks can process them naturally.
I used a variational autoencoder architecture on molecular graphs. The encoder maps a molecular graph to a point in a continuous latent space. The decoder maps a point in that latent space back to a molecular graph. The key is that the latent space is structured: nearby points decode to chemically similar molecules, and the distribution over latent points follows a known prior that you can sample from. Once the VAE is trained, you can do property-conditioned generation by attaching property predictors to the latent space and using Bayesian optimization to find latent points whose predicted properties match your targets, then decoding those points to get candidate molecules.
Training the VAE was the part that took the longest to get right. The standard problem with graph VAEs is posterior collapse, where the decoder learns to ignore the latent code entirely and just generates the most common molecule in the training set. This happens because the KL divergence term in the ELBO loss pushes the encoder toward the prior, which is easy to satisfy if the decoder is flexible enough to reconstruct without needing information from the encoder. The fix is beta annealing: start with a very small weight on the KL term so the encoder can learn a meaningful representation first, then gradually increase it over training so the model learns to compress into the prior without collapsing.
The property predictors are graph neural networks trained on the labeled dataset, predicting Tg, density, and modulus from molecular graph inputs. These run on the decoded molecules to score candidates during the Bayesian optimization step. I used Gaussian processes as the surrogate model for Bayesian optimization because they give calibrated uncertainty estimates, which matters here: you want to explore regions of latent space where the model is uncertain, not just exploit the region around the current best candidate.
The validation setup was: take a vitrimer chemistry I had already simulated with Reformix, remove it from the training set, and ask the inverse design engine to find a molecule with the same properties. The generated candidates got passed to the NNP for fast property evaluation. The best candidates from NNP screening got passed to full Reformix simulation for final validation. In three out of five test cases, the top-ranked generated candidate had simulated properties within 10 percent of the target values.
The failure cases were instructive. When I targeted properties near the edges of the training distribution, outside the range of properties the model had seen during training, the generated molecules were often valid SMILES strings but with chemically implausible bond configurations. The model was hallucinating in the chemistry sense, generating structures that looked reasonable to the VAE but would not actually synthesize. Adding a synthetic accessibility score as an additional constraint during Bayesian optimization reduced this significantly, because it penalizes candidates that are theoretically valid molecules but practically impossible to make in a lab.
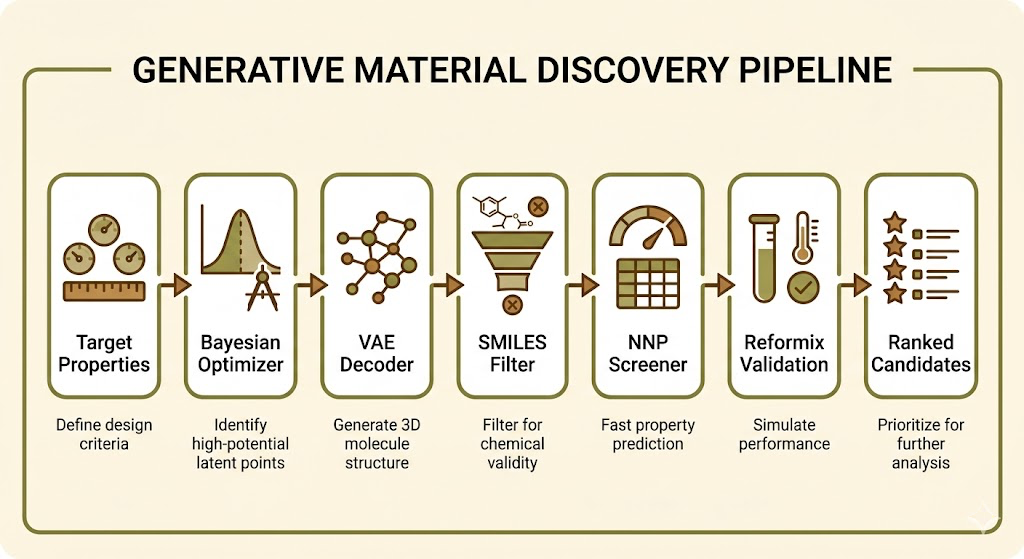
The pipeline now looks like this: specify target Tg, modulus, and density. The Bayesian optimizer searches the latent space to find candidate latent points. These decode to SMILES strings. Invalid SMILES and low synthetic accessibility candidates are filtered out. The remaining candidates get screened with the NNP. The top ten by predicted properties get passed to full Reformix simulation. You get back a ranked list of novel formulations with validated thermomechanical properties, none of which you had to think of yourself.
The goal is to make this the front end of the Reformix screening workflow. A manufacturer specifies what they need: a recyclable adhesive with a Tg between 60 and 80 degrees C and a modulus above 3 GPa. The engine generates candidates, the NNP screens them, Reformix validates the best ones, and the validated blueprints go into the patent pipeline. The human in the loop is making decisions at the business level, not the molecular level.
The part I am still working on is getting the decoder to generate larger polymer repeat units reliably. The current model works well for molecules up to about 50 heavy atoms, which covers simple epoxy-acid systems. Extending it to more complex crosslinker architectures requires either a hierarchical generation approach or a different decoder architecture that handles the longer-range dependencies in larger graphs.