Materials Science · Molecular Dynamics · Deep Tech
2025 — ongoing
Reformix
Designing recyclable polymer at the molecular level.
In 2015, an earthquake killed 9,000 people in Nepal. I was nine years old and I spent the next decade asking why the buildings came down. The answer I kept circling back to was not about engineering failures or corrupt contractors. It was about materials. We built things out of materials that were never designed to last, or fail gracefully, or be replaced. That question about the earthquake eventually led me somewhere I did not expect: recyclable plastics.
Thermoset plastics are some of the strongest materials humans make. Epoxy, carbon fiber, wind turbine blades, the adhesive inside your phone. The reason they are so strong is also why they are a disaster at end of life: once the covalent bonds cure, they are permanent. You cannot melt thermosets. You cannot dissolve them. Every wind blade that reaches end of life, around 8,000 tons of material, goes to landfill. We called it a recycling problem for decades. It is actually a material design problem.
In 2011, a research group at ESPCI Paris synthesized something called a vitrimer. It behaves like a conventional thermoset at room temperature, strong and dimensionally stable, but when heated, the covalent bonds can dynamically exchange. You can remold it, weld it, recycle it. The dead end had an escape hatch. The problem is that designing new vitrimer formulations takes around 18 months of lab trial and error per candidate. The chemical space is enormous and most combinations fail in ways that are hard to predict without just trying them.
So I wanted to simulate the chemistry before synthesizing anything.
This turned out to be harder than it sounds. The key chemical event in vitrimer chemistry, the epoxide ring-opening reaction that forms the crosslinked network, happens on timescales of minutes to hours in reality. Molecular dynamics simulation can access femtoseconds to microseconds. That gap is roughly 15 orders of magnitude. You could run a standard MD simulation for longer than the age of the universe and the reaction would never occur spontaneously.
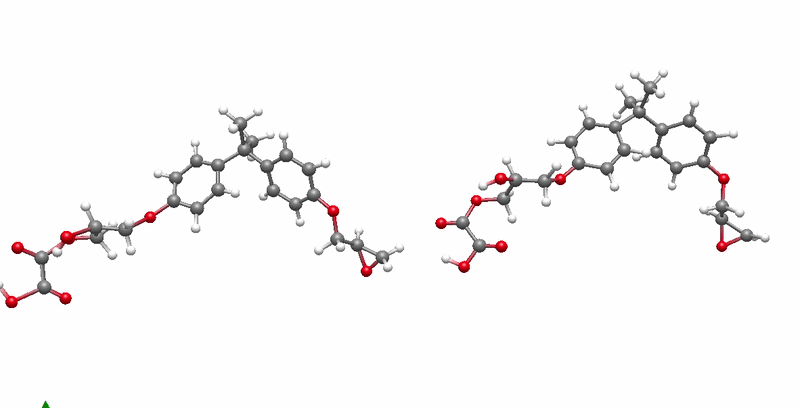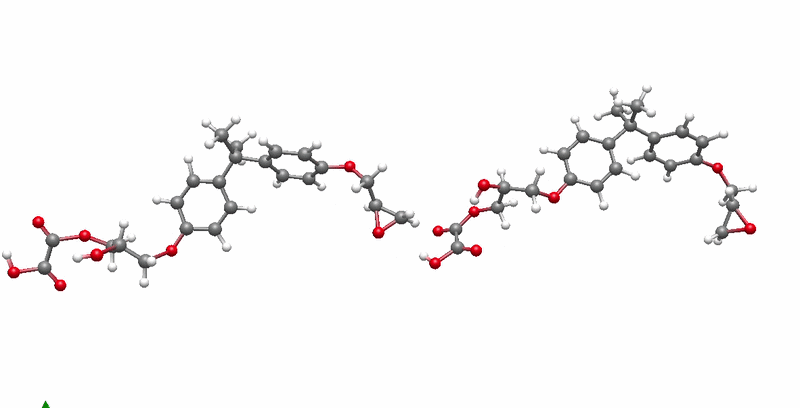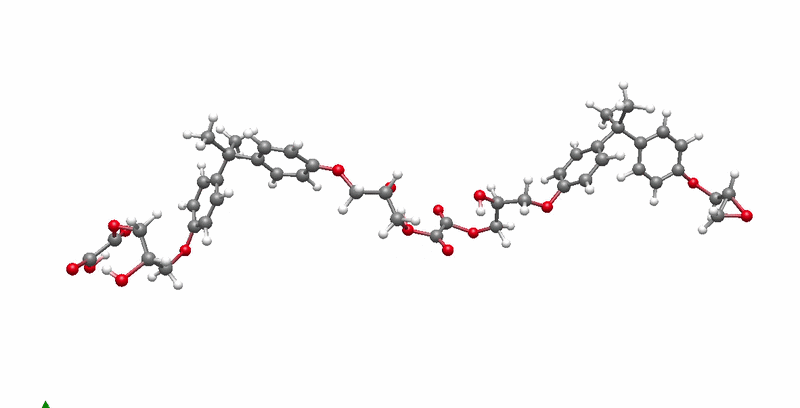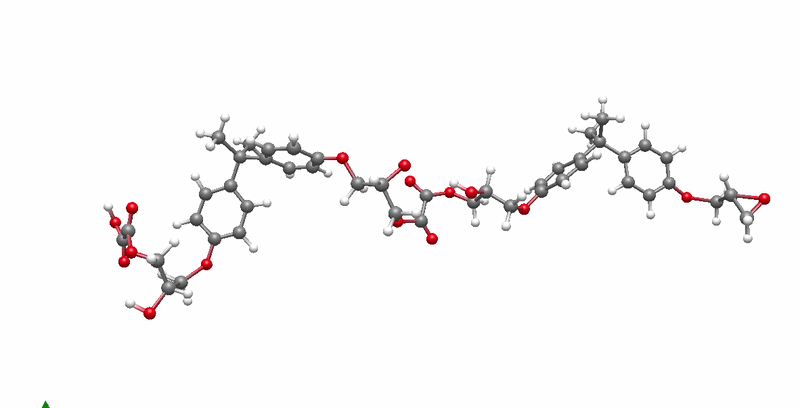
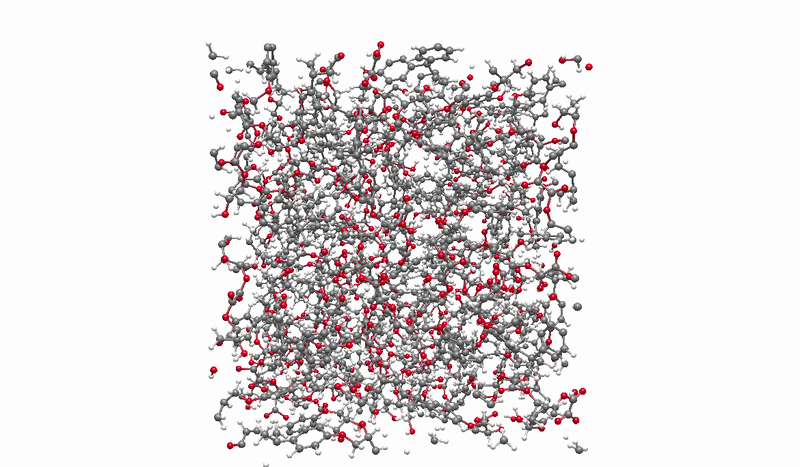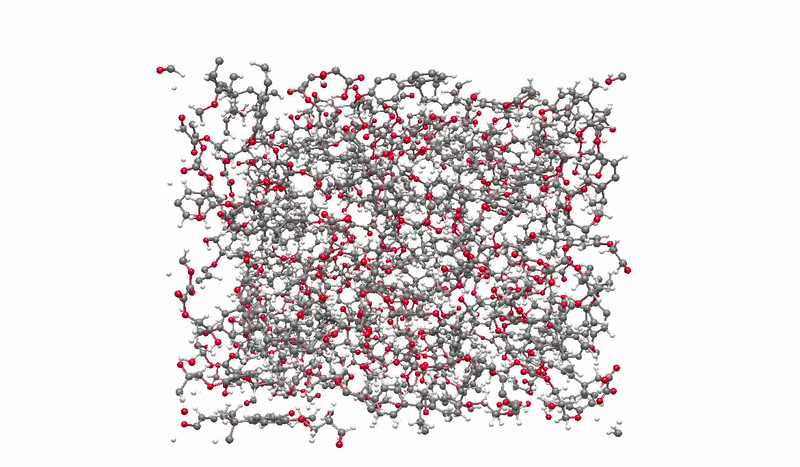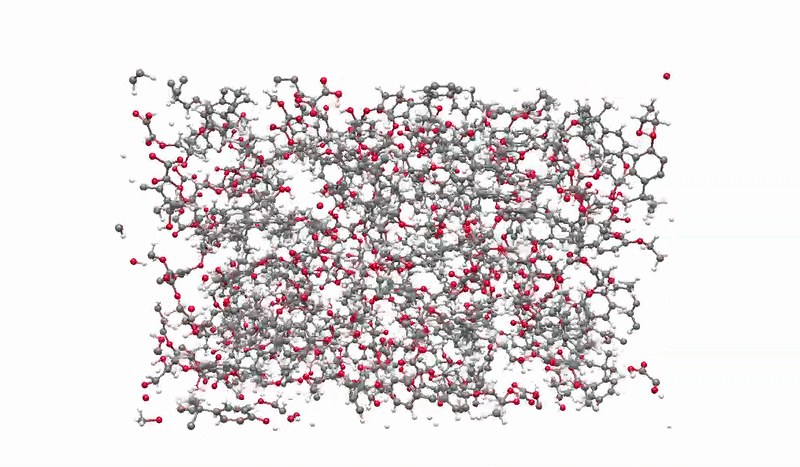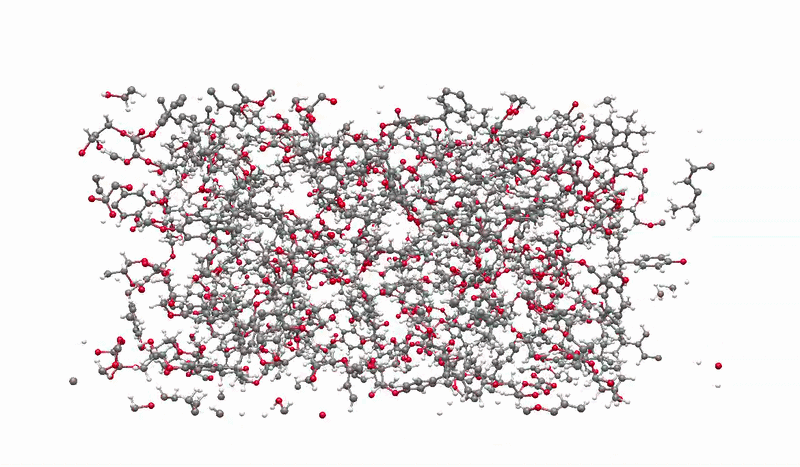
The solution I landed on was biased molecular dynamics using COLVARS inside LAMMPS, paired with a reactive ReaxFF force field that can model real bond breaking and forming at the atomic level. I defined three collective variables to represent the three sequential steps of the reaction: the epoxide C-O bond breaking, the new C-O bond forming between the epoxide carbon and the acid oxygen, and the proton transfer between molecules. A harmonic bias ramps each variable gradually over the course of the simulation, steering the system along the reaction pathway without forcing it unnaturally. This lets the reaction happen on simulation timescales while still capturing the real chemistry.
Before any MD, I pre-optimized all molecular geometries using DFT at the B3LYP/def2-SVP level in ORCA, so the simulation starts from physically realistic structures rather than guesses.
The system I chose to validate this on was DGEBA reacted with oxalic acid. DGEBA is the most widely used epoxy precursor in industry. Oxalic acid is simple and has well-defined reactive sites, which makes it ideal for characterizing the reaction mechanism clearly. No atomistic simulation of this specific system had been published before this work.
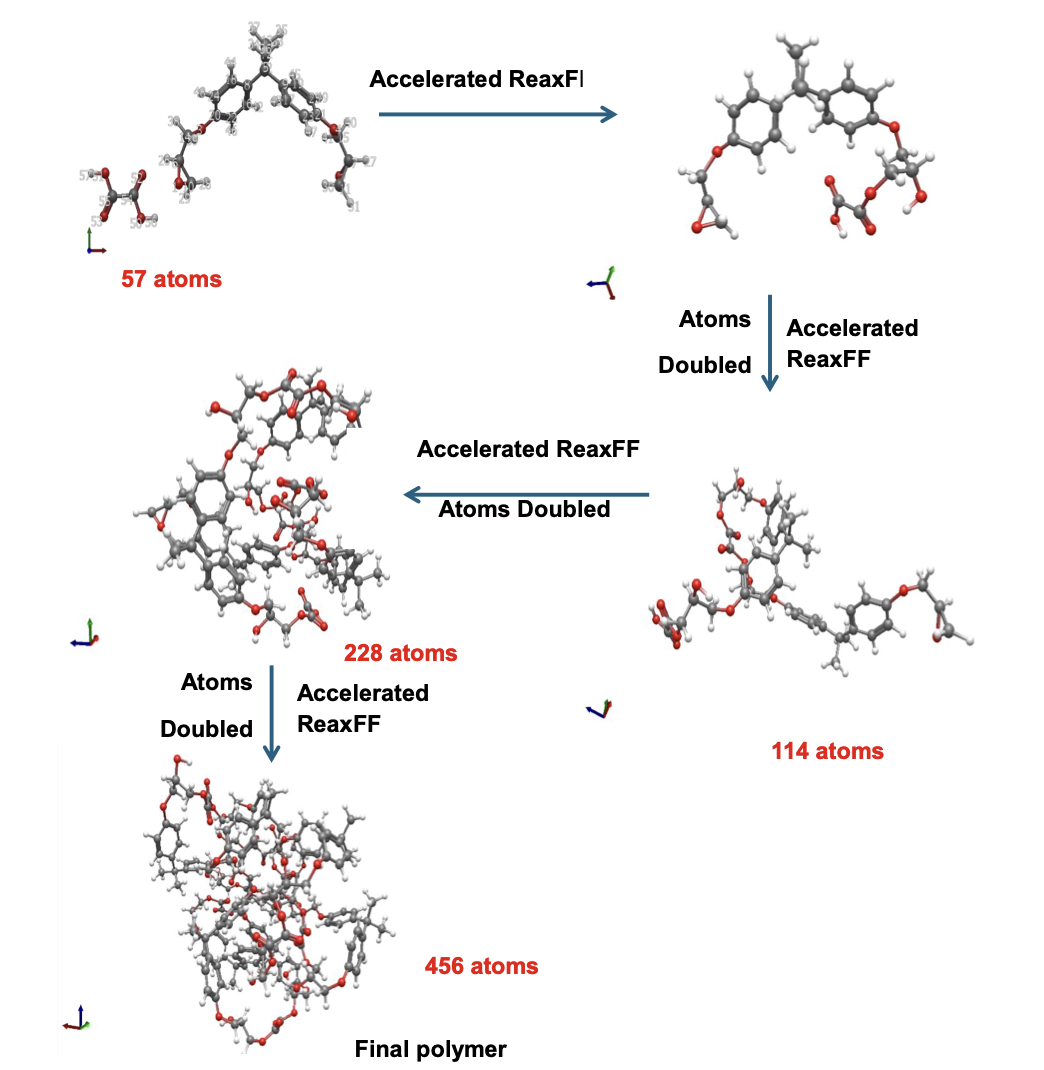
Building the full polymer required a hierarchical approach. I reacted one DGEBA and one oxalic acid molecule under biased simulation, extracted the dimer, re-optimized it with DFT, replicated it, then reacted the doubled system again. I repeated this until reaching a continuous polymer chain of 456 atoms, 8 DGEBA and 8 oxalic acid molecules in 1:1 stoichiometry. Then I packed eight of those chains into a periodic simulation box to approximate bulk behavior, and ran a full annealing protocol from 300K to 600K and back to remove local heterogeneities before measuring any properties.
The simulation took about nine months to get right. A lot of that was debugging runs that produced garbage output, adjusting bias parameters, restarting from scratch, and figuring out why the collective variables were not converging the way they should.
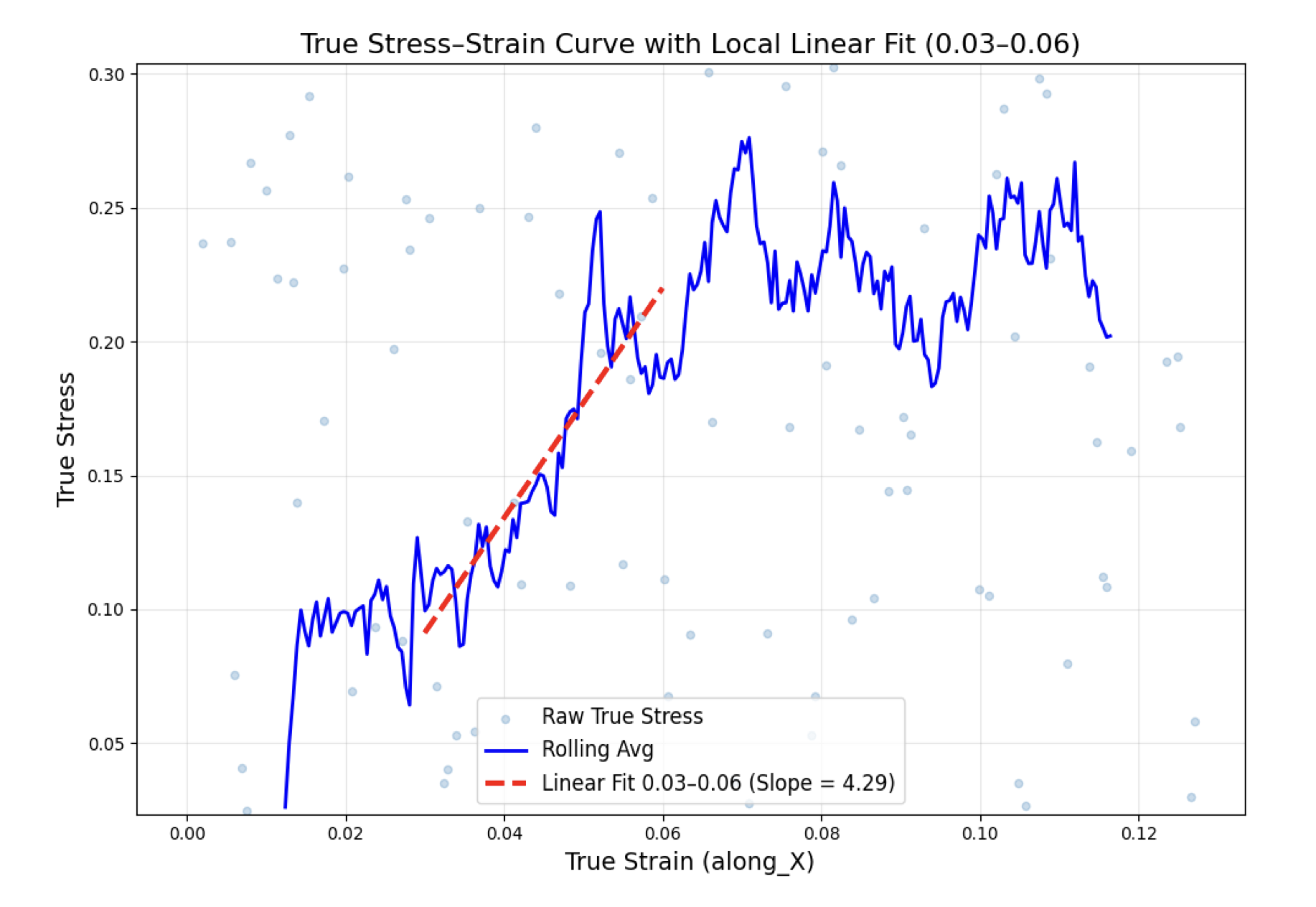
The results, once the engine converged: Density at 300K: 1.2975 ± 0.0009 g/cm³ Glass transition temperature: 76°C Young's modulus: 4.36 ± 1.00 GPa Poisson's ratio: 0.371 ± 0.012
All of these validated against published experimental data for comparable vitrimer systems. The transition state geometry matched DFT predictions. The approach works.
The preprint is on ChemRxiv: doi.org/10.26434/chemrxiv-2026-k6fn6
The broader goal is to use this as a screening engine. Run hundreds of candidate formulations computationally, identify the ones with target properties, patent the validated blueprints, and license them to manufacturers who have production scale but not the molecular design capability. No factory required.
Next: physical synthesis to verify the simulated Tg in a real lab, and a graph neural network layer that predicts material properties from molecular structure in minutes instead of hours, which would make the screening process fast enough for actual industrial use.
Links