Materials · Electrochemistry · Simulation
2025
Solid-State Electrolyte Screener
Finding the battery material that won't catch fire.
Lithium-ion batteries, the ones in your phone and your car and the grid-scale storage unit at the edge of your city, use liquid electrolytes. The liquid is what lets lithium ions shuttle between electrodes fast enough to be useful. It's also what catches fire. The liquid is flammable, it degrades, it leaks. The thermal runaway events that make headlines — battery fires in EVs, in planes, in warehouses full of energy storage — almost always trace back to the liquid electrolyte reaching a temperature it wasn't designed for and turning into a rapid, uncontrolled chemistry problem.
The fix has been known in principle for decades: replace the liquid with a solid. Solid electrolytes don't burn. They have better electrochemical stability windows. They can in theory enable lithium metal anodes, which would nearly double energy density. The problem is finding a solid that conducts lithium ions fast enough to be practical while remaining stable against both electrodes and manufacturable at scale. That combination is extraordinarily rare, and the space of candidate materials is too large to search by experiment alone.
I built a computational screening pipeline to search that space systematically.
Why the solid electrolyte problem is hard
Ionic conductivity in a solid electrolyte is not about the material being a conductor in the electronic sense. It's about lithium ions being able to hop through the crystal lattice or amorphous network from site to site fast enough to sustain meaningful current. That requires a specific combination of structural features: open migration pathways, low activation barriers for the hop, a host lattice that doesn't collapse or phase-separate as ions move through it.
The benchmark for practical solid electrolytes is roughly 1 mS/cm at room temperature. Liquid electrolytes sit around 10 mS/cm. The best solid electrolytes known right now, materials like LGPS (Li₁₀GeP₂S₁₂) and Li₆PS₅Cl argyrodite, approach that benchmark. But they're sulfide-based, which means they react with moisture and they're hard to process. Oxide ceramics like LLZO (Li₇La₃Zr₂O₁₂) are stable but require sintering at 1000°C and have grain boundary resistance issues. The tradeoffs are different everywhere you look, and the vast majority of candidate materials in databases haven't been evaluated for this application at all.
The computational challenge is that ionic conductivity is a dynamical property. You can't compute it from a static crystal structure alone. You have to simulate the atoms moving, the lithium ions hopping, the mean-squared displacement growing over time. That requires molecular dynamics at elevated temperatures, and it requires doing it for dozens or hundreds of candidate materials to get enough signal to make screening decisions.
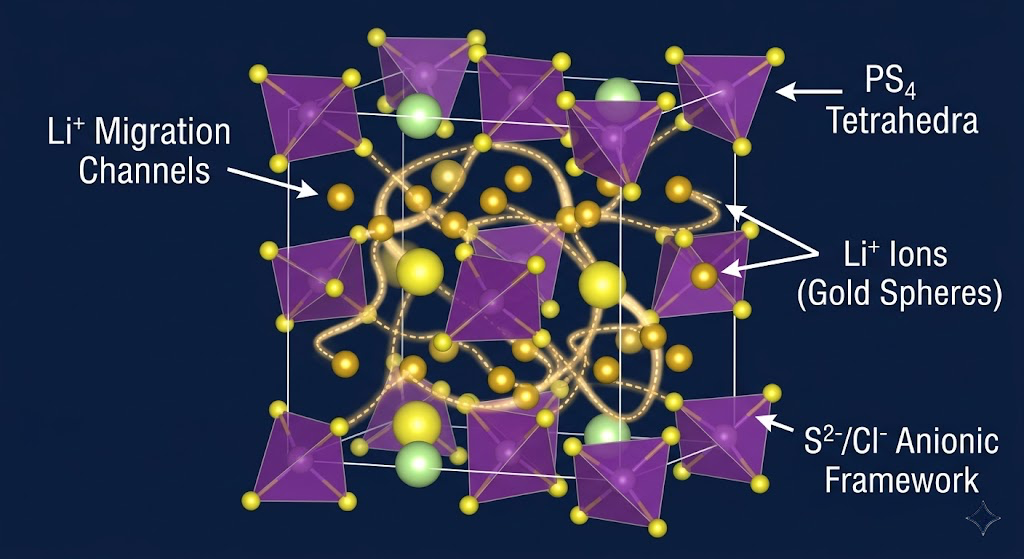
Building the pipeline
The pipeline I built has three stages: candidate selection, property simulation, and ranking.
Candidate selection starts from the Materials Project database, which contains computed structural and electronic data for tens of thousands of inorganic compounds. I filtered for lithium-containing compounds with formal oxidation states consistent with Li⁺ transport, reasonable computed bandgaps (to exclude electronic conductors), and no elements that would be prohibitively expensive or toxic at scale. This gets the candidate pool down to a manageable set of a few hundred materials worth actually simulating.
The simulation stage is where most of the compute happens. For each candidate, I run AIMD using VASP, starting from the relaxed crystal structure from Materials Project. I heat the system to temperatures between 600K and 1200K, which accelerates Li⁺ hopping into a window accessible to nanosecond-scale simulations. I compute the mean-squared displacement of lithium ions over time. Linear MSD growth means diffusive transport. I extract the diffusivity D from the slope and use the Nernst-Einstein equation to extrapolate room-temperature ionic conductivity.
The extrapolation is the most uncertain part. The Arrhenius assumption that log(conductivity) scales linearly with inverse temperature doesn't always hold, especially for materials that undergo phase transitions between your simulation temperature and room temperature. I flag candidates where the Arrhenius fit has high residuals, because those are materials where the temperature extrapolation is least trustworthy and they need closer inspection before drawing conclusions.
In parallel, I run bond valence sum analysis on the crystal structures to map the energetic landscape for Li⁺ migration. BVS gives you a fast approximation of the migration pathway geometry without requiring full AIMD. It's not quantitatively reliable enough to predict conductivity on its own, but it's good enough to identify dead-end sites and bottleneck geometries. When BVS analysis and AIMD agree that a material has good pathway topology, that's a stronger signal than either alone.
The ranking stage combines ionic conductivity, electrochemical stability window (computed from the convex hull decomposition energies in Materials Project), and a crude mechanical stability score derived from the elastic constants available in the database. I weight these into a composite score and sort.
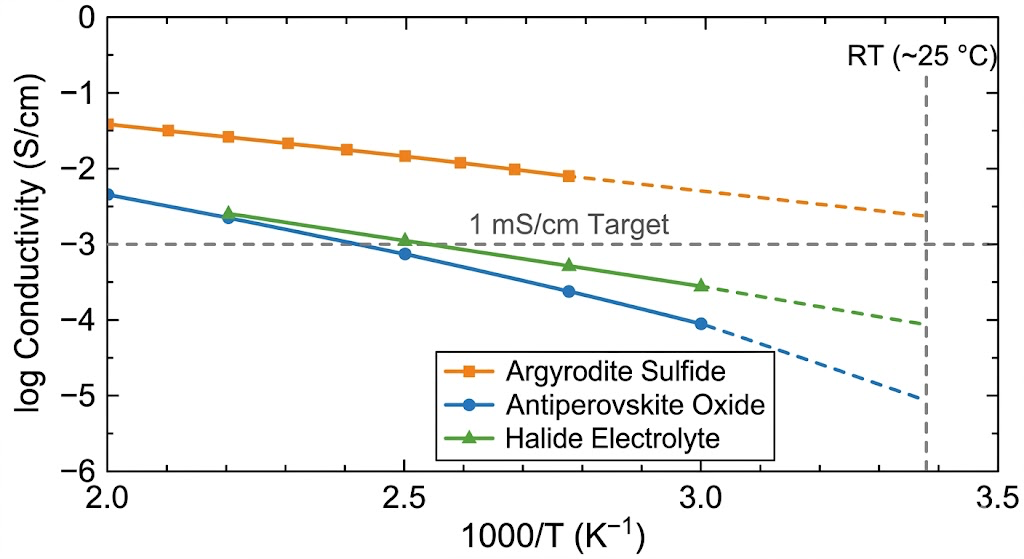
What the simulation taught me
The first thing I learned is how often Materials Project structures are wrong in ways that matter for transport. Small errors in atomic positions or lattice parameters, things that don't affect total energy much, can create artificial bottlenecks in Li⁺ migration pathways. I had to implement a relaxation step before every AIMD run and validate that the starting structure was actually reasonable. Several candidates I was initially excited about dropped out at this stage because their database structures turned out to be approximate or from low-quality DFT runs.
The second thing I learned is that sulfide materials are genuinely better at room temperature than their complexity suggests. The sulfur framework is more polarizable, which means the activation barriers for Li⁺ hopping are lower. The conductivity advantage is real and large, on the order of two to three orders of magnitude over typical oxides. The stability disadvantage is also real. A material that corrodes the moment it contacts moisture is hard to manufacture at scale. The pipeline captures this tradeoff explicitly in the ranking.
The third thing I learned is that the electrochemical stability window calculations from Materials Project, while useful, are often optimistic. They assume thermodynamic equilibrium, and real solid-electrolyte/electrode interfaces are not at equilibrium. They're kinetically stabilized, which means a material that looks unstable by the thermodynamic criterion might still work if the interfacial reaction products form a passivating layer. I added a flag for materials that fail the strict thermodynamic window but where the decomposition products have been reported in the literature to form stable interfaces. These are worth investigating even if the automated score ranks them lower.
The candidates worth pursuing
The pipeline identified three material families as standing out across multiple metrics.
Argyrodite sulfides (Li₆PS₅X, where X is Cl, Br, or I) consistently scored high on conductivity and have reasonable synthetic accessibility despite being sulfide materials. The halogen substitution on the S site opens up additional Li⁺ migration pathways and pushes conductivity above the 1 mS/cm target. The X=Cl composition is the most studied, but my BVS analysis suggests the I-substituted variant might have lower activation barriers through a different pathway geometry that hasn't been experimentally well characterized.
A set of antiperovskite Li₃OX materials (X = Cl, Br) showed up consistently in the top tier on conductivity simulations and have outstanding electrochemical stability windows, because the oxide and halide framework is chemically inert against both lithium metal and standard cathode materials. The challenge is that their ionic conductivity comes with strong anisotropy: it's high along specific crystal directions and much lower perpendicular to them. A pellet of randomly oriented crystallites would average out to a mediocre number. This points to a processing research direction, which is figuring out whether you can texture-align these ceramics during sintering.
The third family is halide electrolytes: Li₃InCl₆ and related compounds. These emerged more recently in the literature and don't have as much database representation, so I had to add them manually. The simulation results are consistent with the experimental reports: good conductivity, excellent oxidative stability, poor reductive stability against lithium metal. They're best suited for cathode-facing electrolyte layers in a composite solid-state architecture rather than as standalone electrolytes.
Where the project is
The pipeline is complete as a research tool. I've validated it against materials with known experimental conductivities, which gives me a sense of the systematic errors in the extrapolation and lets me apply appropriate uncertainty bounds to predictions on unknown materials. The ranking method gives useful ordering even if the absolute numbers are off by a factor of two or three, which is good enough for deciding what to synthesize first.
The obvious next step is collaboration with an experimental group. The value of computational screening is realized when someone actually makes the flagged candidates and measures them. The Li₃OI variant and the texture alignment question for antiperovskites are both specific enough predictions that they're worth a targeted synthesis effort. That connection hasn't happened yet, but the predictions are written down clearly enough that when it does happen, the experimental and computational work can be tightly coupled rather than running in parallel and comparing notes after the fact.
The broader goal is what this kind of tool does to the research process. Right now, most experimental groups exploring solid electrolytes are working from intuition and literature analogy: this compound is similar to a known good one, so let's try it. Systematic screening changes that dynamic. It doesn't replace the experimentalist, but it changes the question they're answering. Instead of "what should I try next?", the question becomes "why does the simulation say this should work, and what does that imply about the underlying physics?" That's a more interesting question.